








Chemicals and reagents used to make DNA reagents for plant genomic DNA isolation (Please visit here for enriched genomic DNA isolation reagents)
CTAB Extraction Buffer
The CTAB buffer mainly includes CTAB, sodium chloride (NaCl), and ethylenediaminetetraacetic acid (EDTA) Tris2-amino-2-hydroxymethyl-1,3-propanediol (TRIS), polyvinylpyrrolidone (PVP), and β mercaptoethanol.
2.1 CTAB
The intricate structure of plant cells, encapsulated within a complex polysaccharide cell wall primarily composed of cellulose, presents challenges for DNA isolation. Cellulose, characterized by its crystalline nature and intermolecular hydrogen bonding, forms a sturdy barrier that must be disrupted to access the genetic material within. Mechanical disruption, often achieved through grinding in the presence of CTAB buffer or liquid nitrogen, weakens the cellulose matrix and facilitates cell wall opening, allowing for subsequent DNA extraction.
Adjacent to the cell wall lies the cell membrane, comprising phospholipid molecules and proteins. These membranes, akin to surfactants, dissolve in detergents possessing amphipathic properties, with hydrophobic tails and hydrophilic heads resembling phospholipid membranes. Among surfactants, ionic variants exhibit superior protein denaturation capabilities, thereby aiding in membrane dissolution.
CTAB, characterized as a cationic detergent, features a lengthy hydrophobic hydrocarbon chain and a hydrophilic head. In aqueous environments during DNA extraction, CTAB interacts with the biological membrane, sequestering lipids and facilitating the release of the nucleus, which lacks a membrane. However, plant tissues, rich in complex polysaccharides and secondary metabolites, may interfere with DNA isolation, necessitating the incorporation of additional compounds such as PVP. Together, CTAB and PVP work synergistically to mitigate the impact of these metabolites, ensuring the successful extraction of pure DNA from plant samples.
Beyond plant cells, CTAB finds utility in DNA isolation from a diverse array of biological sources, including mammalian cells, insect cells, and other living organisms. Its ability to disrupt membranes and sequester lipids makes it indispensable in extracting nucleic acids from various cellular matrices. When compared to alternatives like SDS and PVP, CTAB stands out for its unique cationic nature, which confers distinct advantages in certain applications, particularly in overcoming the challenges posed by complex plant cell walls and secondary metabolites.
We have different strengths of CTAB based extraction buffers are available 2x and 3x. The CTAB can also be purchased separately.
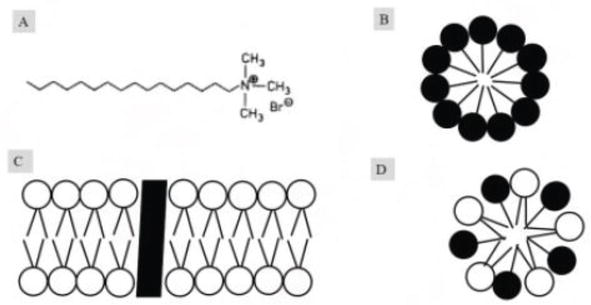
Figure 1.
CTAB’s role in removing membrane. (A) CTAB is amphipathic in nature. It has long hydrocarbon chain (hydrophobic tail) and positively charged trimethylammonium group (hydrophilic head); (B) CTAB forms the micelle into the aqueous solution. Polar heads (hydrophilic) face outside and nonpolar (hydrophobic) hydrocarbon tail hides inside making micelle; (C) biological membrane made up of amphipathic lipids with integral protein; and (D) CTAB captures the membrane lipids and forms the hybrid micelle.
CTAB works differently based on the ionic strength of the solution. At a low ionic strength, it precipitates nucleic acid and acidic polysaccharides (pectin, xylan, and carrageenan), while protein and neutral polysaccharides (dextran, gum locust bean, starch, and inulin) remain in the solution. However, at high ionic concentration, it gets bound to the polysaccharides and forms complexes that are removed during subsequent chloroform extraction. It also denatures or inhibits the activity of proteins and/or enzymes.
2.2 NaCl
Sodium chloride (NaCl) serves multifaceted roles in DNA isolation procedures, contributing to both the precipitation and purification of genomic DNA. During the precipitation step, NaCl effectively neutralizes the negative charges on the phosphate backbone of DNA molecules, facilitating their aggregation and subsequent precipitation from solution—a process commonly referred to as salting out. This aggregation of DNA molecules is crucial for their efficient separation from the aqueous phase, enabling the removal of contaminants and impurities, including proteins, from the DNA sample.
Moreover, NaCl aids in the removal of proteins bound to the DNA by keeping them dissolved in the aqueous layer, preventing their co-precipitation with DNA during alcohol precipitation. By neutralizing the negative charges on the DNA, NaCl promotes the aggregation of DNA molecules while maintaining the solubility of proteins, ensuring their separation from the DNA pellet.
Furthermore, the concentration of NaCl in the DNA extraction buffer influences the efficiency of cell lysis—a critical step in releasing genomic DNA from cellular matrices. Higher salt concentrations, typically above 0.5 M, provide the necessary ionic strength for cetyltrimethylammonium bromide (CTAB) to precipitate polysaccharides, which are abundant in plant cells and can interfere with DNA isolation. Protocols often recommend a NaCl concentration of 1.4 M or higher, especially in procedures designed to eliminate polysaccharides effectively.
Additionally, the osmotic properties of NaCl are integral to cell lysis. In hypotonic solutions, where the salt concentration is lower than that inside the cell, water enters the cell, causing swelling, increased internal pressure, and eventual bursting. Conversely, hypertonic solutions, with higher salt concentrations, lead to water efflux from the cell, resulting in shrinkage and plasmolysis—especially pronounced in plant cells. Therefore, salt concentration plays a pivotal role in achieving efficient cell lysis, ensuring the release of genomic DNA for subsequent isolation and purification.
In summary, NaCl's diverse functions in DNA isolation encompass facilitating DNA precipitation, promoting the separation of DNA from proteins and cellular debris, aiding in cell lysis, and mitigating the interference of polysaccharides, ultimately ensuring the extraction of high-quality genomic DNA suitable for downstream applications in molecular biology research.
Different strengths of NaCl are useful to get a quality DNA from various sources of living materials;
Sodium chloride (NaCl) is a critical component in DNA isolation protocols, and its concentration can significantly impact the efficiency and effectiveness of the process. Different strengths of NaCl are utilized for various purposes in DNA isolation, each serving specific functions tailored to the unique requirements of the sample and the desired outcome. Here are the different strengths of NaCl and their uses in DNA isolation:
Low Concentration (0.15-0.5 M NaCl):
- DNA Solubility: At low concentrations, NaCl is used to maintain the solubility of DNA molecules in aqueous solutions. This concentration range is typically employed in DNA extraction buffers to prevent DNA precipitation before downstream processing steps.
- Protein Dissolution: NaCl at low concentrations helps keep proteins dissolved in the aqueous phase, preventing their co-precipitation with DNA during alcohol precipitation. This facilitates the separation of DNA from proteins and other cellular debris.
Moderate Concentration (0.5-1.5 M NaCl):
- Salting Out: In the precipitation step of DNA isolation, moderate concentrations of NaCl promote the aggregation and precipitation of DNA molecules from solution. This process, known as salting out, is essential for efficiently separating DNA from the aqueous phase.
- Protein Removal: Higher NaCl concentrations enhance the removal of proteins bound to DNA by disrupting protein-DNA complexes. This aids in achieving higher purity of isolated DNA by minimizing contamination with proteins and other cellular components.
High Concentration (Above 1.5 M NaCl):
- Polysaccharide Precipitation: In protocols targeting DNA isolation from plant tissues or other polysaccharide-rich samples, higher concentrations of NaCl are used to precipitate polysaccharides. This helps to minimize interference from polysaccharides, which can hinder DNA extraction and purification.
- Cell Lysis: High salt concentrations are also employed in cell lysis buffers to disrupt cell membranes and release genomic DNA from cellular matrices. This facilitates the efficient extraction of DNA from a variety of biological samples.
Overall, the strategic selection of NaCl concentration in DNA isolation protocols depends on the specific characteristics of the sample and the desired purity and yield of the isolated DNA. By adjusting the NaCl concentration, researchers can optimize DNA isolation procedures to achieve optimal results for downstream applications such as PCR, sequencing, and cloning.
2.3 Tris
Stabilization of pH: Tris buffer acts as a pH stabilizer, maintaining the pH of the DNA extraction solution within a narrow range optimal for enzymatic activity. This is critical for ensuring the effectiveness of enzymes such as nucleases and polymerases, which are used in subsequent steps of DNA isolation.
Minimization of DNA Degradation: Fluctuations in pH can accelerate DNA degradation by nucleases present in the sample. Tris buffer helps to stabilize the pH, thereby minimizing the risk of DNA degradation and preserving the integrity of the isolated DNA.
Enhanced Efficiency of Enzymatic Reactions: Many DNA isolation protocols involve enzymatic reactions such as protein digestion and DNA precipitation. Tris buffer provides an optimal environment for these reactions, maximizing their efficiency and ensuring high yields of pure DNA.
Interference with Polysaccharides: Tris buffer can help to disrupt the complex polysaccharide structures present in plant cell walls, facilitating the release of DNA from the cellular matrix. This is particularly important in DNA extraction from plant tissues, where polysaccharides can hinder the isolation process.
The strength of Tris buffer, typically expressed as its molarity, can impact DNA isolation efficiency in several ways:
Effect on pH: The pH of Tris buffer is dependent on its molarity, with higher molarity solutions exhibiting higher buffering capacity. By selecting an appropriate molarity of Tris buffer, researchers can ensure that the pH of the extraction solution remains within the optimal range for DNA isolation.
Impact on Ionic Strength: The ionic strength of Tris buffer solutions increases with higher molarity. This can affect the efficiency of DNA precipitation and purification steps, as well as the solubility of DNA in solution.
Potential for Contaminant Removal: Higher molarity Tris buffers may facilitate the removal of contaminants such as proteins and polysaccharides from the DNA sample, leading to higher purity DNA isolates.
In summary, Tris buffer is an essential component of DNA isolation protocols from plants, providing pH stability, minimizing DNA degradation, and facilitating enzymatic reactions. The selection of Tris buffer strength influences the efficiency and success of DNA isolation procedures, with higher molarity solutions offering greater buffering capacity and potential for contaminant removal.
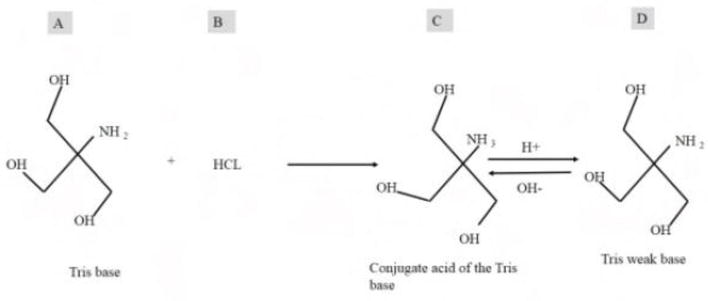
Tris buffer after titration of Tris base solution: (A) with HCL; (B) around pH 8, it contains Tris weak base; (C) its conjugate acid; and (D) in equilibrium it acts as buffer near physiological pH range.
2.4 EDTA
Ethylenediaminetetraacetic acid (EDTA) is a chelating agent commonly used in DNA isolation protocols. Its primary role is to sequester divalent metal ions, such as magnesium (Mg^2+), calcium (Ca^2+), and manganese (Mn^2+), which are cofactors for nucleases and other enzymes that can degrade DNA. By chelating these metal ions, EDTA inhibits the activity of nucleases, thus protecting DNA from enzymatic degradation during the isolation process.
In addition to its role as a nuclease inhibitor, EDTA also helps to maintain the integrity of DNA by preventing the formation of metal-dependent complexes that can cause DNA strand breaks. Furthermore, EDTA can aid in the solubilization of DNA by chelating metal ions that may otherwise interact with DNA and precipitate it out of solution.
Overall, EDTA is an essential component of DNA isolation buffers, where it serves to protect DNA from degradation, maintain its integrity, and promote its solubility. Its ability to chelate metal ions makes it a valuable tool in ensuring the successful isolation of high-quality DNA suitable for downstream applications in molecular biology research and diagnostics.
The EDTA in DNA isolation buffers can significantly impact the efficiency and success of the extraction process, particularly in the context of isolating DNA from plant cells. Different concentrations of EDTA may yield varying outcomes due to their effects on enzymatic activity, metal ion chelation, and overall DNA stability. Here's how different concentrations of EDTA can affect plant DNA isolation:
Low Concentration (0.1-1 mM EDTA):
- Minimal Enzyme Inhibition: Low concentrations of EDTA may provide minimal inhibition of nucleases and other metal-dependent enzymes. While this concentration can help protect DNA from enzymatic degradation, it may not be sufficient to fully inhibit all nucleases present in the plant cell lysate.
- Limited Metal Ion Chelation: At low concentrations, EDTA may chelate only a portion of the metal ions present in the sample. This could lead to incomplete inhibition of metal-dependent enzymes and potential DNA degradation.
Moderate Concentration (1-10 mM EDTA):
- Effective Enzyme Inhibition: Moderate concentrations of EDTA are typically more effective at inhibiting nucleases and metal-dependent enzymes. This can result in better protection of DNA from enzymatic degradation, leading to higher yields of intact DNA.
- Balanced Metal Ion Chelation: At moderate concentrations, EDTA chelates a greater proportion of metal ions, providing more comprehensive inhibition of metal-dependent enzymes. This helps maintain DNA integrity and stability throughout the isolation process.
High Concentration (10-50 mM EDTA):
- Robust Enzyme Inhibition: High concentrations of EDTA offer strong inhibition of nucleases and metal-dependent enzymes, ensuring maximum protection of DNA from degradation. This can be particularly beneficial when working with challenging samples or highly susceptible DNA.
- Complete Metal Ion Chelation: At high concentrations, EDTA effectively chelates nearly all metal ions present in the sample, preventing their interaction with DNA and minimizing the risk of enzymatic degradation. This results in optimal DNA stability and integrity.
Overall, the choice of EDTA concentration in DNA isolation buffers should be carefully optimized based on the specific requirements of the experiment, the susceptibility of the DNA sample to degradation, and the downstream applications. By selecting the appropriate EDTA concentration, researchers can ensure efficient DNA isolation and obtain high-quality DNA suitable for a wide range of molecular biology applications.
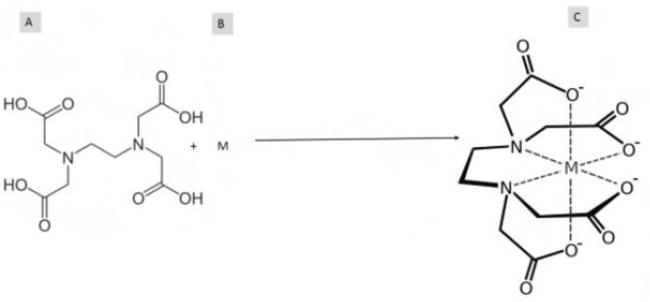
Different strengths of EDTA to be used for various reasons
Higher strengths of ethylenediaminetetraacetic acid (EDTA) in DNA iszotential DNA damage or degradation. This is particularly important when working with precious or limited DNA samples where loss or degradation would be detrimental to the experiment.
Longer Processing Times: In DNA isolation protocols that involve prolonged processing times, such as extended incubation periods or multiple purification steps, higher concentrations of EDTA may be advantageous to maintain DNA stability and integrity over extended durations. This helps ensure that DNA remains protected from enzymatic degradation and metal ion interference throughout the entire isolation process.
Harsh Extraction Conditions: When using harsh extraction conditions, such as high temperatures or chaotropic agents, which can increase the risk of DNA degradation, higher concentrations of EDTA can provide additional protection against enzymatic degradation and metal ion interference. This helps preserve DNA integrity under challenging extraction conditions.
Minimization of Metal Ion Interference: In samples containing high levels of metal ions that could interfere with downstream applications or contribute to DNA degradation, higher concentrations of EDTA are used to effectively chelate metal ions and prevent their interaction with DNA molecules. This ensures the purity and quality of the isolated DNA for subsequent analyses.
Overall, higher strengths of EDTA are employed in DNA isolation when maximum protection of DNA integrity and stability is required, particularly in challenging sample types or experimental conditions where the risk of DNA degradation is elevated. By selecting the appropriate EDTA concentration, researchers can optimize DNA isolation protocols to achieve high-quality DNA suitable for a wide range of molecular biology applications.
Lower strengths of EDTA are also employed In certain experimental setups where the presence of EDTA may interfere with downstream assays or applications. In that case, lower concentrations of EDTA is used. For example, in some enzymatic assays or protein-DNA interaction studies, higher concentrations of EDTA may inhibit enzyme activity or disrupt protein binding, necessitating the use of lower strengths of EDTA.
Figure 3
EDTA chelates divalent cations like magnesium and calcium [25]. (A) Structure of EDTA; (B) “M” depicts the free divalent cations like magnesium and calcium; and (C) EDTA chelates the divalent cations, thereby making unavailable to the DNase and some other activity like cell wall binding and histone-DNA complex formation.
2.5 β-Mercaptoethanol
Plants are rich in phenolics compounds and to get a quality DNA these should be removed. β-Mercaptoethanol (HOCH2CH2SH) is added most of the time in extraction buffers and is a strong reducing agent to clean tannins and other polyphenols present in the crude plant extract. This is added to the extraction buffer just before using the extraction buffer. When it is added try to use fume hood to further process the DNA samples since it gives a very bad smell and it is few of the times harmful when ingested.
Globular proteins get dissolved in water. To make them insoluble, their denaturation is one of the alternatives that can be done at tertiary and quaternary structure level of protein by reducing intermolecular disulfide linkages. β-Mercaptoethanol reduces disulfide bonds of the protein and thus the proteins are denatured.
BME is a reducing agent commonly used in molecular biology and biochemistry applications, including DNA isolation. Its primary function is to break disulfide bonds in proteins, thereby denaturing them and disrupting their structure. This denaturation process is different from the denaturation of proteins by chloroform.
BME Denaturation Mechanism: BME acts by cleaving disulfide bonds (-S-S-) present in proteins, converting them to two sulfhydryl (-SH) groups. This disrupts the tertiary structure of proteins, leading to their denaturation. BME effectively reduces disulfide bonds without causing extensive protein aggregation or precipitation, making it a gentle yet efficient denaturing agent.
Chloroform Denaturation Mechanism: Chloroform, on the other hand, denatures proteins through a different mechanism. It disrupts protein structure by solubilizing the lipid components of cell membranes and organelles, leading to membrane disruption and protein release. Chloroform's denaturation effect is primarily attributed to its ability to dissolve lipids and disrupt cellular membranes, rather than directly affecting protein structure through chemical modification.
Use in DNA Purification: The difference in denaturation mechanisms between BME and chloroform is leveraged in DNA purification protocols to obtain pure genomic or nuclear DNA free from protein contamination. BME is commonly used in DNA extraction buffers to denature and solubilize proteins present in cell lysates, allowing for selective isolation of DNA. By breaking disulfide bonds in proteins, BME ensures that proteins are fully denatured and remain soluble in the extraction buffer, while DNA remains intact.
Protein-Free DNA Isolation: After denaturation with BME, chloroform is often used in DNA extraction protocols to separate the DNA-containing aqueous phase from the protein and lipid-containing organic phase. Chloroform's ability to disrupt cell membranes and solubilize lipids facilitates the partitioning of DNA into the aqueous phase, while proteins and lipids are retained in the organic phase. This enables the isolation of pure, protein-free genomic or nuclear DNA suitable for downstream molecular biology applications such as PCR, sequencing, and cloning.
In summary, BME denatures proteins by breaking disulfide bonds, while chloroform disrupts protein structure by solubilizing lipids and disrupting cell membranes. These differences in denaturation mechanisms are exploited in DNA purification protocols to obtain pure DNA free from protein contamination, with BME facilitating protein denaturation and solubilization, and chloroform enabling the separation of DNA from protein and lipid components.
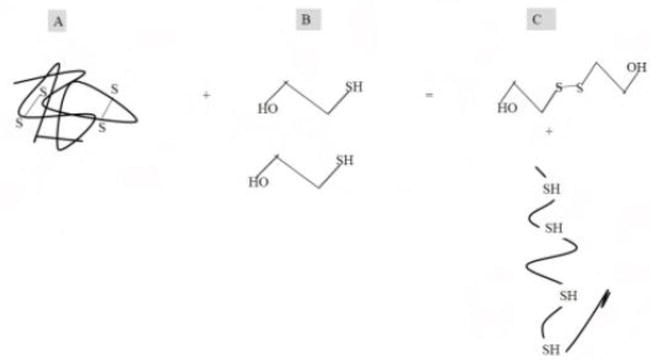
Figure 4.
β-Mercaptoethanol reduces disulfide linkage of protein, thus denaturing it [25]. (A) Protein tertiary structure with disulfide bonds; (B) β-mercaptoethanol; and (C) oxidized β-mercaptoethanol and protein denatured by β-mercaptoethanol via its ability to cleave disulfide bonds.
2.6 PVP
PVP is added to remove phenolic compounds from plant DNA extracts. Polyphenol is a major component in medicinal plants, woody plants, and mature plant parts. It is present in the vacuole, while its oxidizing enzyme, polyphenol oxidase (PPO) is located in plastid [15]. During grinding of the tissue, compartmentalization breaks and PPO convert polyphenols into quinone, which gives brown coloration. Polyphenols bind DNA and make downstream processing difficult as they get co-precipitated with the nucleic acid. PVP removes polyphenolic contamination by binding it through hydrogen bond [16, 17]. Thus, it prevents polyphenol oxidation, and thereby browning of DNA samples [18]. When the extract is centrifuged with chloroform, PVP complexes get accumulated at the interphase.
Cell lysate mixture with CTAB buffer should be kept in the water bath at 65°C, which irreversibly inhibits enzyme DNase. After removing the sample from water bath, it should be allowed to cool at room temperature, then chloroform:isoamyl alcohol (24:1) or phenol:chloroform:isoamyl alcohol (25:24:1) shall be added. Chloroform:octanol (24:1) can also be used instead of chloroform:isoamyl alcohol (24:1).
3. Phenol
Phenol is an organic solvent, so it is not miscible with water and is used along with chloroform and isoamyl alcohol for purification of the DNA to remove proteins and polysaccharide contaminants. When phenol is shaken with cell extract, the nonpolar components of the cell will be fractionated in phenol, leaving polar ones in water. DNA is insoluble in phenol because phenol is a nonpolar solution. On the other side, protein has both polar and nonpolar groups present in it because of the long chain of different amino acids. Different amino acids have different groups present on their side chain. Also, the folding of the protein into the secondary, tertiary, and quaternary structure depends on the polarity of the amino acids. The bonds between amino acids are broken by the addition of phenol and protein gets denatured and ultimately the protein becomes unfolded.
Centrifugation after phenol:chloroform:isoamyl alcohol in 25:24:1 ratio steps gives three layers, that is aqueous, interphase, and at bottom organic phase. At neutral to alkaline pH, the nucleic acids are negatively charged and polar. Therefore, it is hydrophilic and remains in an aqueous phase. In aqueous solution, hydrophobic amino acid forms a protective core. However, after denaturation, nonpolar cores (hydrophobic) get exposed, causing precipitation of protein as well as some polysaccharides at interphase.
The phenol-chloroform combination reduces the partitioning of poly (A) and mRNA into the organic phase and reduces the formation of insoluble RNA protein complexes at the interphase. Phenol retains about 10–15% of the aqueous phase, which results in a similar loss of RNA; chloroform prevents this retention of water and thus improves yields.
Only neutral phenol should be used, as acidic phenol dissolves DNA within, or phenol turns into quinones by oxidation and it forms free radical, degrading nucleic acid. Simple observation of phenol’s pink color will state its acidic nature. The centrifugation after chloroform:isoamyl alcohol step should be done under room temperature, because below 15°C, CTAB/nucleic acid forms irreversible aggregates and may precipitate. During this step, the DNA shall be in aqueous phase [19].
4. Chloroform
Chloroform disrupts proteins by solubilizing the lipid components of cell membranes and organelles, leading to membrane disruption and protein release. This degradation occurs through the following mechanisms:
Disruption of Cell Membranes: Chloroform is a lipophilic solvent, meaning it has an affinity for lipids. When added to a cell lysate or homogenate, chloroform penetrates the lipid bilayer of cell membranes and organelles, disrupting their integrity. This disruption releases proteins from their native cellular compartments into the surrounding solution.
Solubilization of Lipids: Chloroform solubilizes lipids, including phospholipids and cholesterol, which are major components of cell membranes and organelle membranes. By dissolving these lipids, chloroform disrupts the structural integrity of cell membranes, leading to their fragmentation and the subsequent release of proteins.
Partitioning of Proteins: As chloroform disrupts cell membranes and solubilizes lipids, proteins become partitioned into the aqueous phase of the solution. This partitioning occurs due to the hydrophilic nature of proteins, which allows them to remain in the aqueous phase while lipids are extracted into the organic phase.
Denaturation and Aggregation: Chloroform exposure can also induce denaturation of proteins, altering their secondary and tertiary structures. Additionally, chloroform-induced membrane disruption can expose hydrophobic regions of proteins, leading to their aggregation and precipitation.
Chloroform's ability to degrade proteins and disrupt cellular structures is important in DNA isolation for several reasons:
Protein Removal: Proteins can interfere with DNA isolation procedures by binding to DNA or contaminating DNA samples. Chloroform effectively removes proteins from cell lysates or homogenates, allowing for the isolation of pure DNA.
Cell Lysis: Chloroform aids in the lysis of cells by disrupting their membranes, releasing cellular contents including DNA, proteins, and organelles. This facilitates the extraction of DNA from cells and tissues.
Separation of DNA: Chloroform is often used in DNA extraction protocols to separate the DNA-containing aqueous phase from the protein and lipid-containing organic phase. This partitioning allows for the isolation of pure DNA free from protein and lipid contaminants.
Overall, chloroform plays a crucial role in DNA isolation by facilitating cell lysis, protein removal, and the separation of DNA from other cellular components. Its ability to degrade proteins and disrupt cell membranes is essential for obtaining high-quality, protein-free DNA suitable for downstream molecular biology applications.
5. Isoamyl alcohol
Chloroform comes in contact with the air and forms gas phosgene (COCl2, carbonyl chloride), which is harmful. If we simply use chloroform only, the gas entrapment causes foaming or frothing, it foams up between interphase during extraction process and makes it difficult to properly purify the DNA, which is prevented when chloroform is used along with isoamyl alcohol or isopentanol {(CH3)2CHCH2CH2OH} or octanol {CH3(CH2)7OH} by preventing the emulsification of a solution. Isoamyl alcohol or isopentanol is not miscible in the aqueous solution because it is a long-chain aliphatic compound, containing five carbon atoms and stabilizes the interphase between organic and aqueous layer. The aqueous phase contains DNA and the organic phase contains lipid, proteins, and other impurities. Isoamyl alcohol helps to inhibit RNase activity and to help prevent the solubilization in the phenol phase of long RNA molecules with long poly (A) portions. This will increase the purity of DNA.
6. Ribonuclease A
Genomic DNA should be treated with Ribonuclease A (RNase A) to remove the contamination of RNA for DNA purification. RNase A is an endoribonuclease that catalyzes the hydrolysis of the 3′,5′-phosphodiester linkage of RNA at the 5′-ester bond in a two-step reaction. The first step is a transphosphorylation to give an oligonucleotide terminating in a pyrimidine 2′,3′-cyclic phosphate. The second is the hydrolysis of the cyclic phosphate to give a terminal 3′-phosphate. Numerous chemical studies have suggested that histidine 12, histidine 119, and lysine 41 are involved in the active site of the enzyme and the DNA is devoid of 2′OH group (deoxy), it remains secure (Figures 5 and 6) [20].
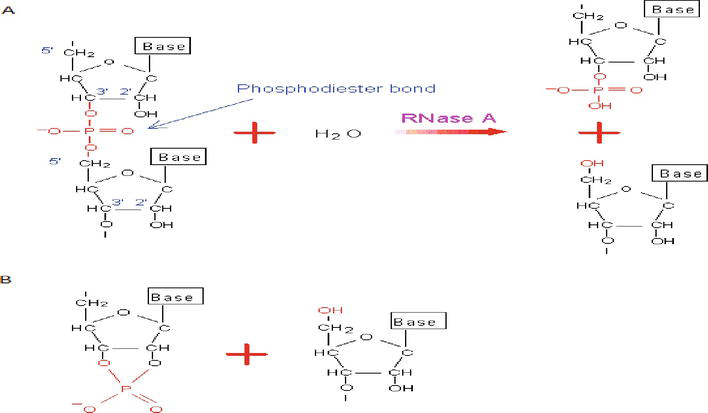
Figure 5.
(A) The hydrolysis reaction catalyzed by RNase A. An RNA molecule is a chain of nucleotides linked by the phosphodiester bond, which may be cleaved by RNase [27]. (A) This figure shows only two nucleotides adjacent to the cleavage site and (B) the intermediate product (transition state) of this reaction.
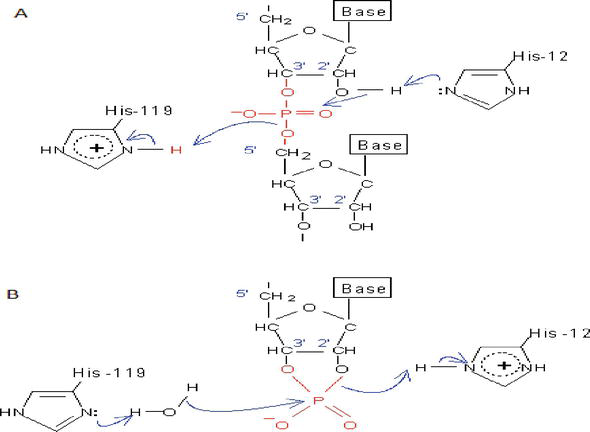
Figure 6.
The catalytic mechanism of RNase A, which contains two critical residues: His-12 and His-119. (A) The transition state is formed by electron transfer from His-12 to His-119, passing through 2′-OH and (B) after the transition state is formed, the electron can move from His-119 to His-12, generating the final product. DNA lacks the critical 2′-OH and thus cannot be catalyzed by RNase A.
7. Isopropanol/ethanol
Alcohol is used to precipitate the DNA out of the extraction solution, so we can wash all those salts and chemicals away and then dissolve it in our final solvent—usually water or some variant of Tris-EDTA solution. DNA remains dissolved in aqueous solution because DNA has phosphodiester backbone, which is hydrophilic in nature. Water molecule forms hydration shell around DNA by forming hydrogen bonds. Isopropanol/ethanol is used in precipitation of DNA, which breaks the hydration shell. Isopropanol is a good choice for precipitation of DNA. The amount of isopropanol requirement is less (0.6–0.7 volume of supernatant), as isopropanol has a higher capacity to reduce the dielectric constant of water than the ethanol (2–3 volume) and also requires a fair amount of salt to work. RNA which has extra 2′OH remains hydrogen bounded with water more strongly than DNA tends to stay soluble in it, thus selective precipitation of DNA can be done. Isopropanol also dissolves nonpolar solvents such as chloroform, thus the impurities form previous step can also be removed.
Using ice-cold isopropanol is generally practiced, but many researchers say that it should be used at room temperature, otherwise it will precipitate polysaccharides also [21]. Though the yield of DNA will be increased at low temperature, it may increase impurities.
8. Sodium acetate/ammonium acetate/potassium acetate/sodium chloride/lithium chloride/potassium chloride
The role of the salt in the extraction protocol is to neutralize the charges on the sugar phosphate backbone of the DNA. Sodium acetate with pH 5.2 is commonly used for precipitation of nucleic acid along with ethanol [23]. In solution, sodium acetate dissociates into Na+ and [CH3COO]−. The positively charged sodium ions neutralize the negative charge on the PO3− groups on the sugar phosphate backbone of nucleic acids reducing repulsion between DNA molecules, making the DNA molecule far less hydrophilic, and therefore much less soluble in water. The electrostatic attraction between the Na+ ions in solution and the PO3− ions on the nucleic acid are dictated by Coulomb’s Law, which is affected by the dielectric constant of the solution. Water has a high dielectric constant, which makes it fairly difficult for the Na+ and PO3− to come together. This is useful in aggregation and formation of tangled mass. It is also called as salting out. Nevertheless, it is not seen when salt alone is used. It requires the solution with low dielectric constant, which allows this interaction. This is affected by either ethanol or isopropanol, which has a much lower dielectric constant, making it much easier for Na+ to interact with the PO3−, shield its charge, and make the nucleic acid less hydrophilic, causing the DNA to drop out of solution (Figure 7).

Figure 7.
Role of salt in DNA precipitation [25]. (A) DNA molecules in aqueous solution have the negative charge and repel each other; (B) sodium acetate dissociates into the water into sodium and acetate ion; and (C) sodium ion shields the negative charge on the DNA molecules by neutralizing it and helps in aggregation and precipitation.
9. Ethanol
DNA precipitate is washed again with 70% ethanol to rinse excess salt that might come along with the extraction buffers from the pellet, centrifuged, and ethanol is discarded, leaving DNA in the precipitate. Precipitate is air-dried or vacuum-dried. Over drying should be avoided as DNA converts B form to D form, which is difficult to dissolve later.
10. Tris-EDTA (TE) buffer/sterile water
In older times in DNA isolation methods, DNA used to be stored dry and diluted when required. Nowadays, for long-term storage, it is prudent to store DNA in a buffer that maintains its pH and keeps it from getting degraded. TE buffer contains Tris (10 mM) and EDTA (1 mM), where Tris is the buffering component and EDTA the chelating component. For DNA isolation, the pH is usually set to 7.5–8.5, the slight alkalinity of TE buffer also prevents chances of acid hydrolysis that may further disrupt the stability of DNA stored in water. Tris amino constituent of TE buffer has the ability to protect DNA strands from radiation damage, in both solid state and fluid solution. As radiation produces free radicals, it may break DNA strands. Thus, in the fluid solution at ambient temperature Tris acts by scavenging hydroxyl radicals [26]. The purpose of EDTA is to chelate Mg2+ ions in solution necessary for DNase or RNase action, thus protecting the DNA from DNases or RNase.
Sterile water can be utilized for short-duration storage of DNA. If TE buffer is used for storage of DNA, it should be diluted further with sterile water to dilute EDTA concentration for making magnesium ions available for polymerase activity during PCR because if DNA has to be sent for sequencing afterward, the buffer components in TE hinders the process. The same EDTA that chelates ions to degrade magnesium also hinders the action of DNA polymerases during PCR, which can be overcome by adding more magnesium to the master mix, or perhaps diluting the DNA sample so that the already low concentrations of EDTA do not actually disrupt PCR. In fact, in a large number of cases, they do not.
Important notice: The content has been pasted here as it is from a research article belongs to ina Heikrujam, Rajkumar Kishor and Pranab Behari Mazumder DOI: 10.5772/intechopen.92206. The article available at Heikrujam, J., Kishor, R., Mazumder, P. B. , 2020, 'The Chemistry Behind Plant DNA Isolation Protocols', in O. Boldura, C. Baltă, N. S. Awwad (eds.), Biochemical Analysis Tools - Methods for Bio-Molecules Studies, IntechOpen, London. 10.5772/intechopen.92206.
We claim no copy rights for the content from this article. The publisher can write to us if they wishes to delete the content from here